Dr. Debasis Maity
Consultant Uro-Surgeon
Medical Discussion
Renal Cystic Diseases
Causes:
Inheritable
-
Autosomal dominant
Q- ADPKD (adult variety)
- Medullary cystic disease
- Familial Hypoplastic Glomerulocystic Disease
- Multiple malformation syndromes with renal cysts (Tuberous Sclerosis, VHL)
-
Autosomal recessive
Q- ARPKD (infantile variety)
- Juvenile nephrophthisis
- Congenital nephrosis (familial nephrotic syndrome)
Non-inheritable
- Multicystic dysplastic kidney
- Benign multilocular cyst (cystic nephroma)
- Medullary sponge kidney
- Sporadic Glomerulosclerotic Kidney Disease
- Acquired renal cystic disease
- Calyceal diverticulum (pyelogenic cyst)
Types:
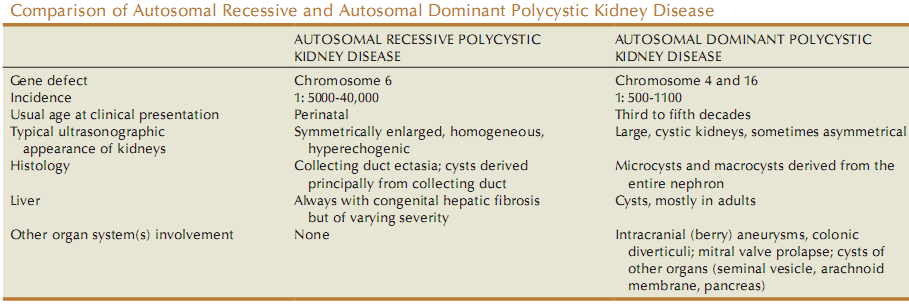
Autosomal Dominant Polycystic Kidney Disease (ADPKD)
REMEMBER:
-
Epidemiology:
- Incidence is 1:500-1000 live birth.
- Becomes clinically apparent after 30 yrs but may present in utero.
-
Genetics:
Gene involved Chromosome Protein Function of normal gene PKD-1 16 Polycystin-1 Inhibition of cellular proliferation PKD-2 4 Polycystin-2 Inhibition of cellular proliferation -
Clinical memoranda:
- Cysts can be identified sonographically before age 20 years in almost all affected individuals.
- No higher incidence of RCC in this population.
PATHOLOGY:
Must know
-
Renal cysts (few mm to few cm): Appear diffusely throughout cortex and medulla with communications at various points.
-
Earliest finding in fetus is focal tubular dilatation
Q, occurs anywhere along the nephron. -
Associated anomalies:
-
Cysts of liver (m/c), (F>M; appears later than renal cyst), pancreas, spleen, lungs
-
Berry aneurysm
Q(10-40% cases; leads to SAH and death in 9%) -
Aortic aneurysm
-
Colonic diverticula
- Mitral valve prolapse
Q
-
Good to know
-
Other pathologies:
-
Adenoma formation in the cyst wall
-
Arteriosclerosis
-
Interstitial fibrosis due to infection and inflammation
- Apoptosis in the cyst epithelial lining may lead to renal failure
-
Q: Why there is renal failure in ADPKD although only 1% of all nephron are involved?
A: Probably the following factors play their roles:
- Compression of non-dilated nephron by the cyst
- Apoptosis in epithelial cell lining of cysts
- Secondary effects of hypertension
PRESENTATION:
-
In utero: due to large cystic kidney, it may present as respiratory distress or still birth.
-
Typical features if age of presentation is >1 y:
- Hematuria
- Proteinuria
- Hypertension
-
Usual age of presentation is 30-50 y. In this age group, typical presentations are:
- Hypertension (principle form of presentation)
- Microscopic and gross hematuria (without as such anemia) in 50%
- Renal colic due to formation and passage of clots or stones in 50-70% cases (20-30% of all patients develop stones)
- Flank pain
- GI problems
- Hepatic cysts
- Berry aneurysm
EVALUATION:
Diagnosis is presumed if:
-
H/O renal disease, hypertension and stroke in 3 generations
OR,
-
If no familial history, then:
B/L renal cyst
+
Two or more of the following is present:
-
3 or more hepatic cysts
-
Cerebral artery aneurysm
- Solitary cyst of pineal gland, arachnoid matter, spleen, pancreas
-
Diagnostic Modalities:
-
USG
- Shows renal cysts + cysts in different organs
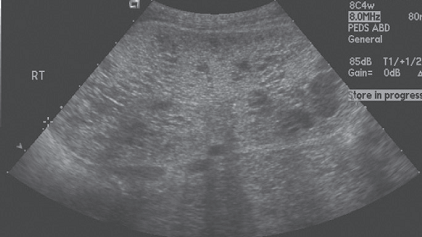
- Shows renal cysts + cysts in different organs
-
IVU
Reveals:
- B/L renal enlargement
- Calyceal distortion
- “Bubble or Swiss cheese appearance”
Qon nephrogram
-
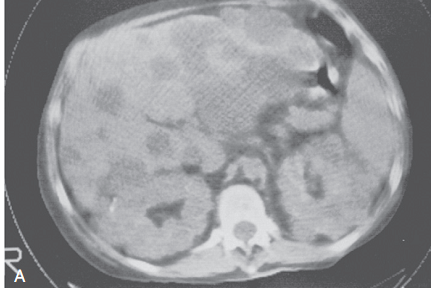
CT
- Superior to USG
- Detects acute hemorrhage in cysts (50-90 Hansfield unit)
-
MRI
- Indicated in the patient of renal insufficiency.
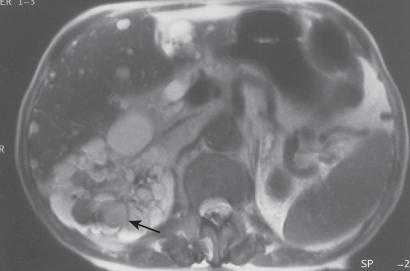
- Indicated in the patient of renal insufficiency.
-
Cytogenetic study
Reveals defects in chromosome 16 and less frequently in chromosome 4
-
Overnight water deprivation test
Decreased urine Molalitymax (680 +/- 14 mosm)
-
Amniocentesis
In utero evaluation of the cause of cystic renal disease (as picked up by different imaging modalities) to be ADPKD, ARPKD or Tuberous Sclerosis
TREATMENT:
After diagnosis at presymptomatic phase (diagnosed accidentally/prenatally)
- To monitor with RFT and BP measurement
- Advantages of diagnosis at this stage:
- Ability to prevent HTN/UTI
- To identify potential kidney donor
- To offer advice on marriage or childbearing
- To provide prenatal diagnosis in the next generation
After diagnosis at symptomatic phase
- Definitive treatment
- Percutaneous aspiration with or without instillation of sclerosing agents like alcohol.
- Symptomatic treatment
- Pain relief is done by deroofing the cyst (Rovsing’s operation).
- Treatment of complications
- UTI is seen in females. 87% involve cysts, 91% involve parenchyma. Infections are ascending in nature, mostly.
- Managed with lipid soluble antibiotics like FQs, Chloramphenicol, cotrimoxazole
Terminal phase (48% patient reaches ESRD by the age of 73 y)
Dialysis and/or Renal transplant
Emerging therapy:
- EGF and its receptor play a role in pathogenesis of ADPKD.
- EGFR-TK inhibitor is used successfully in mice.
Autosomal Recessive Polycystic Kidney Disease (ARPKD)
REMEMBER:
-
Epidemiology:
- Incidence is 1:40,000 live births
- If not apparent at birth, disease will be apparent in late childhood upto 13 y of age.
-
Genetics:
Gene Chromosome Protein Effect of mutation PKHD-1 Ch. 6 Fibrocystin/Polyductin Fibrocystin is abundant in normal fetal kidney collecting duct but absent in ARPKD affected kidney. -
Clinical memoranda:
- Earlier the age of presentation, more severe the disease.
- All patients present with hepatic fibrosis
Q. - Who survives first 31 days, good chance to survive for at least 1 year more.
PATHOLOGY:
- B/L large cystic kidney
- All patients have liver involvement as the following forms:
- Hepatic fibrosis
- Billiary ectasia
- Periportal fibrosis
- Younger the patient, milder the liver disease. Also, younger the patient with hepatic fibrosis, milder the renal cystic disease.
PRESENTATION:
-
Oliguria is the rule (h/o oligohydramnios may be there in mother)
-
Infants present as:
- A non-transilluminating kidney shaped non-bosselated flank mass
- Display Potter’s facies, deformity of limbs, respiratory distress due to pulmonary hypoplasia
- Initially, serum urea and creatinine is similar to the mother due to the dialysis by placenta during in-utero period, but, soon rise.
- If presented in birth, patient dies within 2 months due to uremia and respiratory failure.
- Children present as:
- Hypertension
- Renal failure
EVALUATION:
-
USG:
Reveals –
- Very enlarged, homogeneously hyperechogenic kidneys, when compared with liver echogenecity.
-
Pyramids are also hyperechoic.
- Comparative features on USG picture of different lesions of kidney.
Condition Feature on USG ADPKD Cysts are diffuse and large ARPKD Hyperechoic homogeneous enlarged kidneys Multicystic dysplastic kidney Hypoechoic cyst in a non-reniform mass ,with little parenchyma Wilm’s tumor Heterogeneous mass with functioning kidney Renal vein thrombosis Renal enlargement with hypoechoic medulla 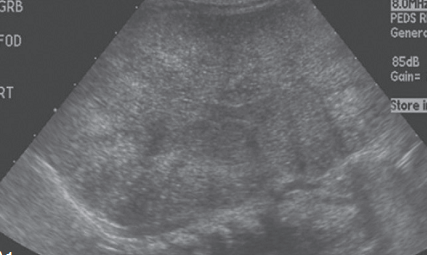
-
CT scan:
- More sensitive
- Macrocyst are rare
- Cysts <1cm more often appear
- Diffuse hyperechoic foci without shadow-composed of calcium oxalate/citrate
-
IVU:
- Radial/medullary streaking (sunburst pattern) caused by dilated collecting tubules filled with contrast.
- Genetic counselling
?
TREATMENT:
- No cure.
- Those who survive require treatment for hypertension, CCF, renal and hepatic failure.
- Portal hypertension is dealt with Warrant Shunt.
? - Treatment of oesophageal varices, if needed.
- Haemodialysis/Renal transplantation in some patients.
OTHER DISEASES CAUSING RENAL CYSTS
Juvenile Nephrophthisis (JN) / Medullary Cystic Disease complex (MCD)
- Chromosome 2 involved
- Clinical hallmark for these diseases: polyuria, polydypsia and hypertension
- Histologically show identical features: severe interstitial nephritis and cysts at cortico-medullary junction.
- JN manifests in children (10-20% of all renal insufficiency in children) which leads to ESRD by early teenage.
- MCD manifests in early adulthood.
Multiple malformation syndromes with renal cysts
Tuberous sclerosis (TS)
-
Genes involved:
- TSC1 (chr. 9)
- TSC2 (chr. 16)
-
Triad:
- Epilepsy + mental retardation + adenoma sebaceum
- Associated anomalies:
- Renal angiomyolipomas (40-80%)
- Renal cysts (20%)
- RCC (2%)
von Hippel-Lindau (VHL)
- VHL gene on Ch. 3 is involved
- VHL is a tumor suppressor gene involved in regulation of hypoxia-inducible factor (HIF) target genes, including VEGF abnormal or absent VHL leads to increased tissue levels of specific growth factors and, in particular, VEGF, the most prominent growth factor in RCC.
- Associated anomalies:
- Cerebellar and retinal hemangioblastoma
- Cysts of kidney (76%), pancreas and epididymis
- Pheochromocytoma (10-12%)
- Clear cell RCC (50%)
-
Types:
Type 1: without pheochromocytoma
Type 2: with pheochromocytoma
- 2A: without RCC or pancreatic cyst
- 2B: with RCC and pancreatic cyst
- 2C: Familial pheochromocytoma without hemangioblastomas and RCC